
Focus sur une pathologie
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
Les myosites, encore appelées myopathies inflammatoires, sont une grande famille hétérogène de maladies auto-immunes dont le dénominateur commun historique est l’atteinte du muscle strié squelettique. Avec les progrès des connaissances, ce groupe a pu être subdivisé en 4 principales familles de myosites dont les principales atteintes sont cutanées, articulaires et pulmonaires. Les myosites sont des maladies chroniques appartenant à la famille des connectivites et dont la prise en charge globale a été radicalement améliorée pendant les dernières années avec le progrès des connaissances, mais dont l’évolution naturelle et le pronostic reste très variable en fonction du type de myosite.
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
Epidemiologie
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
LES TYPES DE MYOPATHIES INFLAMMATOIRES
Quels sont les différents types de myosite ?
Les myosites sont actuellement subdivisées en 4 sous-groupes :
– Les dermatomyosites, qui sont caractérisées par une éruption cutanée typique (description ci-dessous).
– Les myosites nécrosantes autoimmunes, qui sont caractérisées par une atteinte musculaire (description ci-dessous) au premier plan alors que les signes extra musculaires sont rares.
– Les syndromes de chevauchement, qui sont caractérisés par des signes extra-musculaires et des signes cutanés différents des signes de dermatomyosites.
On distingue:
o Le syndrome des anti-synthétases défini par l’association d’au moins un des signes suivants (le plus souvent au moins deux signes sont présents) : une polyarthrite, un syndrome de Raynaud, une pneumopathie, une myosite, des mains de mécaniciens (description ci-dessous), une fièvre ET un anticorps dirigés contre les amino-acyl-ARNt synthétases (description ci-dessous).
o La scléromyosite, associant des signes de myosite et des signes de sclérodermie (description de cette connectivite dans une rubrique dédiée).
o La lupomyosite, associant des signes de myosite et de lupus.
– Les myosites à inclusion sont caractérisées par un début le plus souvent après 45 ans, une faiblesse musculaire des fléchisseurs des doigts et des quadriceps ainsi qu’une absence de réponse aux traitements immunomodulateurs conventionnels.
Chacun de ces 4 groupes est ainsi caractérisé par un syndrome clinique spécifique, la présence d’anticorps spécifiques (description ci-dessous) et une réponse thérapeutique particulière (description ci-dessous) témoignant d’une physiopathologie particulière à chacun des sous-groupes
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
MANIFESTATIONS CLINIQUES
Quels sont les symptômes des myosites ?
Les myosites sont des maladies systémiques dont les symptômes associent à des degrés divers des manifestations dermatologiques, articulaires, musculaires, pulmonaires et vasculaires.
La présence de tous les symptômes n’est pas nécessaire pour évoquer le diagnostic, qui repose sur un faisceau d’arguments cliniques mais également paracliniques.
Chacun de ses symptômes peut inaugurer de façon isolée les myosites.
Ainsi, même si l’on parle de myosite, il faut avoir à l’esprit que l’atteinte musculaire peut être entièrement absente (formes amyopathiques) ou être au 2ème plan (forme hypomyopathique) de la maladie.
Les manifestations dermatologiques sont un signe phare de la dermatomyosite et des syndromes des anti-synthétases.
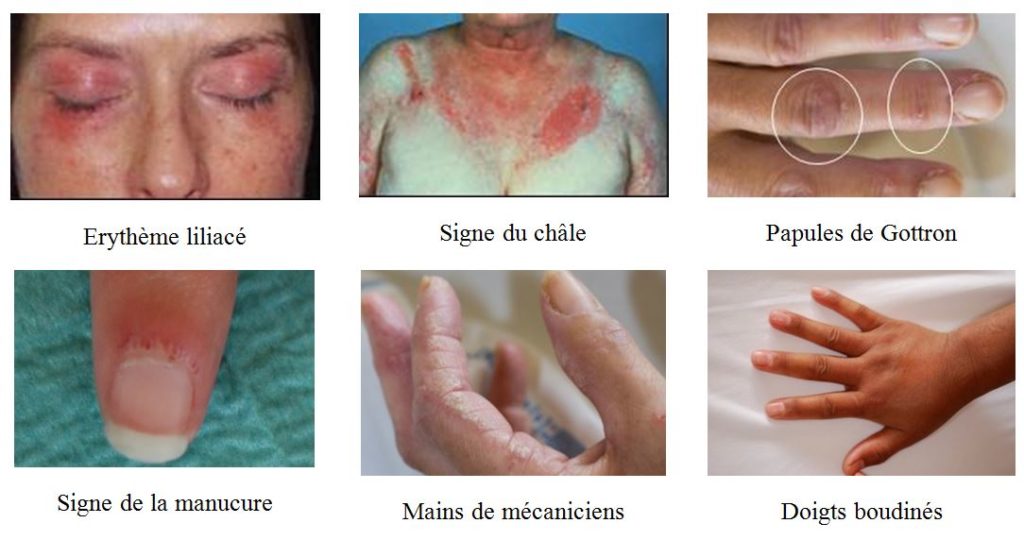
Les signes cutanés spécifiques de dermatomyosite (photo) comprennent :
o Les papules de Gottron, qui sont des papules érythémateuses ou violacées apparaissant sur la face d’extension des doigts, des coudes ou des genoux
o L’érythème en bandes du dos des mains et pieds, dont la différence avec celui du lupus se fait par le fait qu’il repose sur les articulations (alors qu’il se situe entre les articulations en cas de lupus)
o Le signe de la manucure est un érythème inflammatoire squameux à proximité du lit de l’ongle souvent accompagné de dilatation et d’une tortuosité des petits vaisseaux sanguins (capillaires) de cette région, qui est visible à l’examen à l’œil nu (ou à la capillaroscopie)
o L’érythème liliacé des paupières rappelant la couleur de l’héliotrope, pouvant concernant également le nez mais épargnant alors sa pointe et son arrête
o Le signe du châle est une éruption érythémateuse confluente touchant le haut des épaules et la nuque
o L’érythème en « V » : érythème confluent autour de la base antérieure du cou et du haut de la poitrine,
o Le signe du holster est un érythème touchant la face latéral des cuisses.
o La présence de lésions cutanées nacrées enchâssées sur les plis de la face palmaire des mains est évocatrice de dermatomyosite à anticorps anti-MDA5
Les lésions de « mains de mécanicien » sont un épaississement hyperkératosique parfois fissuré concernant la face latérale des doigts. Ce signe se rencontre surtout au cours du syndrome des antisynthétases
La présence d’une sclérose cutanée et/ou d’un aspect de doigts boudinés se rencontre surtout au cours des scléromyosites.
Une calcinose cutanée est une complication tardive des myosites
Manifestations musculaires
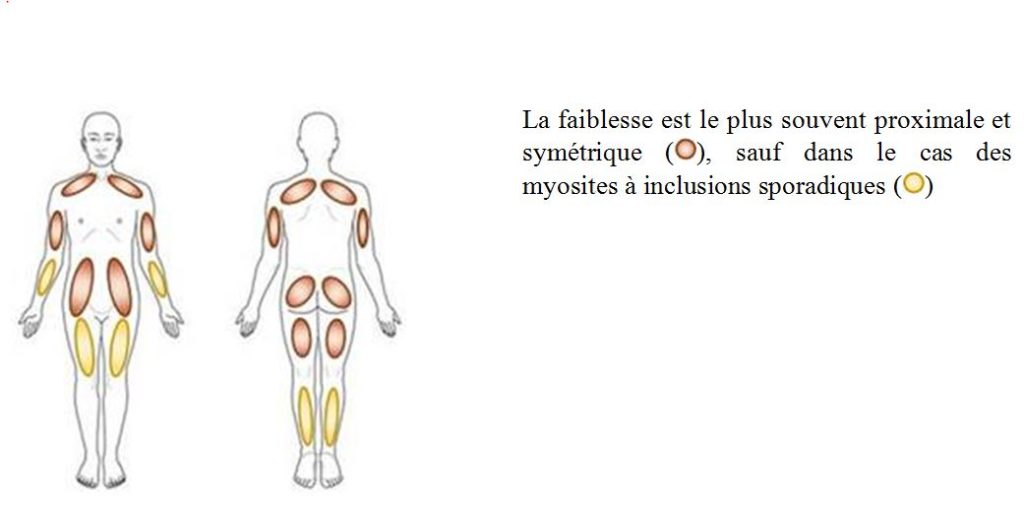
Elles consistent en une faiblesse bilatérale et symétrique des muscles prédominant à la racine des membres supérieurs et inférieurs. L’importance de cette faiblesse est très variable allant d’une limitation modérée lors des efforts importants à une impossibilité à mouvoir les bras et/ou les cuisses.
Une faiblesse prédominant sur l’extension des jambes (muscle quadriceps) et la flexion des doigts est caractéristique des myosites sporadiques à inclusion.
Des douleurs musculaires peuvent être présentes mais ne sont pas constantes (et ne sont pas spécifiques des myosites).
Il peut parfois exister une atrophie des masses musculaires (amyotrophie).
L’atteinte des muscles responsables de la déglutition peut entrainer dysphagie, de fausses routes qui doivent conduire à une prise en charge plus intense.
L’atteinte du muscle cardiaque est rare mais grave.
Biologiquement, il existe le plus souvent une élévation du taux sanguin de créatine kinases (CK ou CPK).
La faiblesse est le plus souvent proximale et symétrique, sauf dans le cas des myosites à inclusions sporadiques.
L’électro-neuro-myogramme est utile pour exclure une faiblesse en rapport avec une atteinte nerveuse (atteinte du motoneurone) ou une atteinte de la jonction entre le nerf moteur et le muscle (jonction neuromusculaire).
Cet examen mais souvent en évidence des signes de souffrance musculaire (syndrome myogène).
L’IRM musculaire peut aussi mettre en évidence des anomalies musculaires (œdème, dégénérescence graisseuse atrophie). Ces signes ne sont cependant pas spécifique des myosites.
Manifestations articulaires
Il s’agit le plus souvent d’arthralgie d’horaire inflammatoire des petites articulations.
Parfois il existe une polyarthrite qui ressemble à la polyarthrite rhumatoïde. Une authentique polyarthrite (avec anticorps anti-CCP) peut être associée aux myosites. L’atteinte est classiquement sans dégâts ostéo-articulaires sur les radiographies. Cependant des signes radiologiques peuvent se rencontrer.

Manifestations pulmonaires
Les manifestations pulmonaires se rencontrent surtout au cours du syndrome des anti-synthétases, de la scléromyosite et de la dermatomyosite à anticorps anti-MDA5.
Il s’agit d’une dyspnée, de gravité variable (allant d’une dyspnée pour les efforts soutenus au syndrome de détresse respiratoire aigu). L’apparition d’une dyspnée rapidement progressive constitue un signe de gravité.
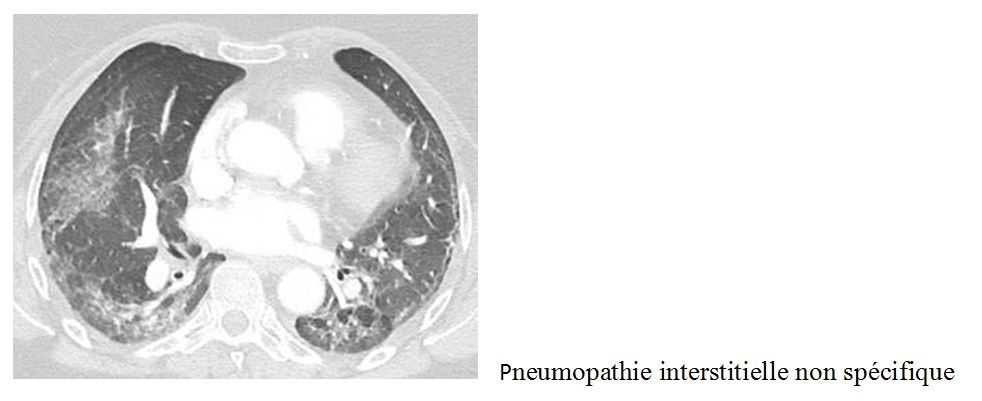
Le scanner thoracique permet le diagnostic. Il s’agit le plus souvent de lésions de pneumopathie interstitielle non spécifique, mais des lésions de pneumopathie interstitielle commune, de pneumopathie organisée, de bronchiolite ou encore d’atteinte alvéolaire diffuse peuvent être observés.
Les explorations fonctionnelles respiratoires sont utiles pour quantifier l’atteinte et suivre son évolution.
Manifestations vasculaires
La capillaroscopie est utile pour mettre en évidence cette atteinte.
Le phénomène de Raynaud est fréquent dans les myosites.
Des ulcérations pulpaires d’origine vasculaire sont rares mais peuvent se voir surtout dans la scléromyosite,
Une hypertension artérielle pulmonaire se rencontre surtout au cours des scléromyosites.
Manifestations générales
Un amaigrissement et une asthénie sont souvent présents.
Une fièvre, un syndrome inflammatoire sont fréquents aux cours des myosites de chevauchement et des dermatomyosites. Ils sont rares au cours des autres myosites.
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
DIAGNOSTIC
Des critères de diagnostic et classification des myosites ont été proposés par l’ACR/ EULAR 2017. Ces critères peuvent aider le clinicien à porter un diagnostic et permettent de créer des groupes homogènes de patients dans les études. Cependant, ces critères ne prennent en compte qu’un signe extra-musculaire de myosite (l’éruption de dermatomyosite) et un seul auto-anticorps (l’anti-Jo1) ce qui explique que des patients atteints de myosite peuvent ne pas répondre à ces critères (en particulier les patients atteints de myosite de chevauchement non-anti-Jo1). D’autre part les pièges diagnostics sont nombreux.
Le diagnostic doit donc être suspecté devant les signes expliqués plus haut et confirmé dans un centre expert
Le diagnostic de myosite repose en pratique sur un trépied diagnostic:
Signes cliniques, décrits ci-dessus
Hormis l’éruption de dermatomyosite, aucun des signes décrits ci-dessus n’est spécifique de myosite. La présence d’un des signes doit faire rechercher les autres signes. L’association des signes augmente la probabilité du diagnostic.
Recherche d’autoanticorps:
Les myosites sont des pathologies auto-immunes. Une vingtaine d’autres auto-anticorps ont montré leurs utilités pour le diagnostic et la classification des myosites. Pourtant les critères ACR/EULAR ne prennent en compte que les anti-Jo1 pour le diagnostic des myosites.
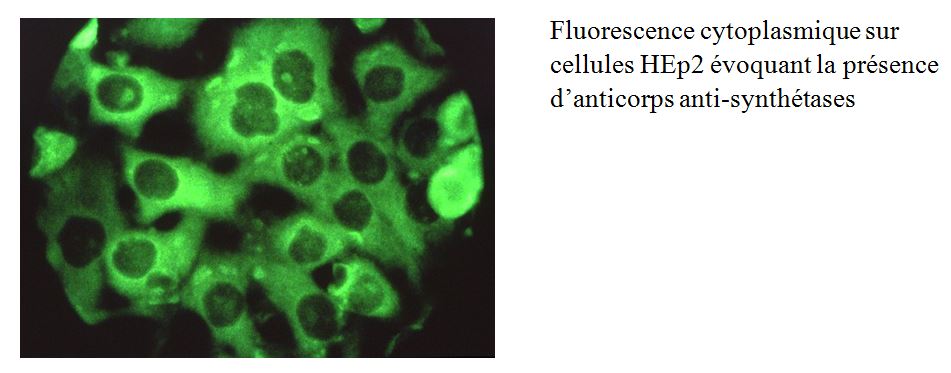
La recherche de ces auto-anticorps nécessite une expertise importante dans ce domaine. Plusieurs kits commerciaux existent dont les performances diagnostiques diffèrent en fonction des auto-anticorps. Certains laboratoires en France ont développés des techniques « maison ». La confrontation des résultats avec l’immunofluorescence sur cellules HEp2 et avec la clinique est importante pour interpréter le résultat.
Les anticorps associés aux myosites :
o Dermatomyosites : anti-Mi2, -TIF1-γ, -NXP2, -SAE, -MDA5
o Syndrome des anti-synthétases : anti-Jo-1, -PL7, -PL12, -OJ, -EJ, -KS, -Ha, -Zo
o Myopathies nécrosantes auto-immunes : anti-SRP, -HMGCR
o Scléromyosite : anti-U1-RNP, -PM/Scl, -Ku, -centromère -Scl70, -Th/To, -U3-RNP
Chez environ 20% des patients atteints de myosites, il n’y a pas d’auto-anticorps mis en évidence (ces patients ont probablement des auto-anticorps qui ne sont pas encore découverts ou pour lesquels il n’y a pas de technique de recherche disponible en pratique courante).
La biopsie musculaire:
La biopsie musculaire consiste en un prélèvement de tissu musculaire squelettique, parfois de son fascia, en vue de son examen histologique et parfois biochimique et moléculaire. La biopsie musculaire « à ciel ouvert » est la technique la plus courante en France.
Elle est un élément crucial du diagnostic de myosite. En plus de sa valeur diagnostique, la biopsie musculaire aide à classifier le type de myosites et pourrait apporter des éléments pronostiques. Pour plusieurs experts, devant une myopathie, lorsque des signes extra-musculaires de myosites sont présents (en particulier l’éruption de dermatomyosite) et qu’ils sont associés à un anticorps spécifique de myosite, une biopsie musculaire n’est pas indispensable au diagnostic. Cependant, même dans le cas de la dermatomyosite, la biopsie musculaire augmente la performance diagnostic.
Elle doit être réalisée dans un centre expert maîtrisant les étapes nécessaires à l’exploitation diagnostique du matériel : prélèvement d’une quantité suffisante de tissus, congélation immédiate dans l’isopentane, réalisation d’une dizaine de colorations. L’interprétation des lésions nécessite une grande expérience. En particulier, la présence d’infiltrat inflammatoire n’est pas synonyme de myosite et se rencontre aussi au cours de certaines myopathies génétiques. A l’inverse, l’absence d’infiltrat inflammatoire n’exclut pas le diagnostic et est une caractéristique des myopathies nécrosantes auto-immunes.
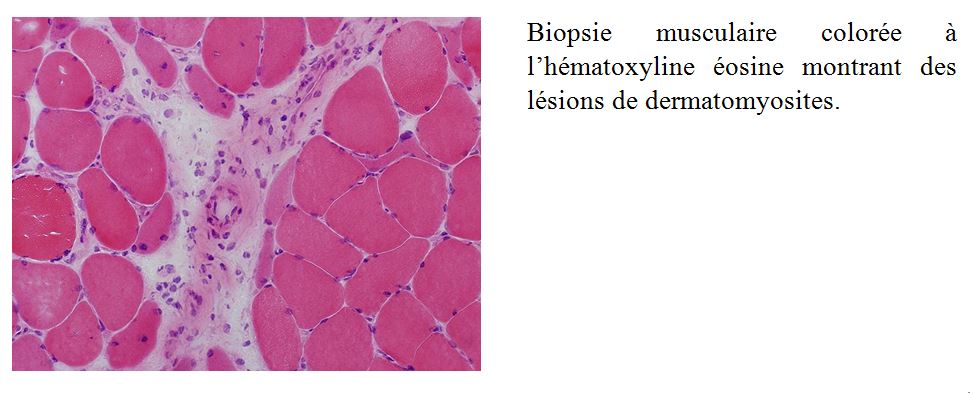
Les diagnostics différentiels
Les diagnostics différentiels sont nombreux, tout comme les pièges diagnostics en fonction de la présentation et du contexte diagnostic :
o Les autres affections neuromusculaires périphériques : le déficit moteur périphérique doit faire éliminer les pathologies du nerf périphérique et de la jonction neuromusculaire qui peuvent s’accompagner d’une myolyse modérée et d’anomalies de l’imagerie musculaire. Une myolyse franche doit faire rechercher les autres causes de myopathies. Un infiltrat inflammatoire à la biopsie musculaire se rencontre dans des pathologies génétiques.
o Les pneumopathies interstitielles non-autoimmunes :les pneumopathies interstitielles ont de nombreuses causes et les origines médicamenteuses, infectieuses, environnementales, spécifiques et génétiques doivent être considérées.
o Les autres rhumatismes inflammatoires : les autres rhumatismes inflammatoires peuvent être pris à tort pour une myopathie inflammatoire lorsqu’il existe des myalgies, un œdème musculaire à l’IRM et des signes extra-musculaires communs aux myosites (pneumopathie interstitielle, syndrome de Raynaud).
Les éruptions cutanées :
o Les mains de mécaniciens peuvent être confondues avec d’autres causes de pulpites des doigts (irritatives et allergiques).
o L’érythème liliacé des paupières peut être confondu avec des pathologies beaucoup plus fréquentes (eczéma de contact, dermatite atopique, dermatite séborrhéique, rosacée).
o Les macules et papules de Gottron peuvent être confondues avec le psoriasis.
o L’érythème en zone photo-exposée peut être confondu avec un lupus
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
TRAITEMENTS
Le traitement doit être débuté le plus rapidement possible
Les objectifs du traitement sont :
1. Rechercher des signes de gravités,
2. Evaluer le risque, rechercher et traiter un cancer associé,
3. Contrôler l’activité de la maladie,
4. Limiter et reverser les dégâts de la maladie,
5. Prévenir les comorbidités.
Rechercher des signes de gravités,
Il s’agit des signes suivants :
o Perte de la marche,
o Dysphagie compromettant l’alimentation,
o Myocardite,
o Atteinte respiratoire sévère.
Evaluer le risque, rechercher et traiter un cancer associé,
Dans leur ensemble, les myosites sont associées à un sur-risque de cancer. Le risque de cancer est multiplié par environ 5 en cas de dermatomyosite et par environ 2 pour les autres myosites par rapport à la population générale.
Cependant, ce sur risque est présent dans les 3 ans qui suivent le diagnostic de myosite. Passé ce délai le risque rejoint celui de la population générale.
Plusieurs sous-groupes de myosites ne sont pas associés à un cancer : les myosites de l’enfant, les myosites de chevauchement, les myosites à inclusions, certaines dermatomyosites et certaines myosites nécrosantes autoimmunes.
Ainsi, la stratégie de recherche d’un cancer dans les 3 ans qui suivent le diagnostic de myosite doit être adaptée au risque de cette association.
Quand un cancer est associé, son traitement permet de traiter aussi la myosite.
Contrôler l’activité de la maladie,
Les myosites sont des maladies chroniques évoluant dans la plupart des cas par des poussées suivies de périodes de rémission. Certaines formes de myosites sont associées à des poussées initiales uniques, et peuvent parfois guérir. Le traitement dépend du type de myosite et de sa gravité.
La corticothérapie est prescrite initialement (phase d’induction de la rémission) à forte dose (par voie orale à 1 mg/kg/j et parfois en bolus). Ce traitement à un délai d’action rapide mais expose à long terme à un risque de nombreux effets secondaires, raison pour laquelle la corticothérapie est sevrée progressivement jusqu’à son arrêt.
Les immunoglobulines intraveineuses sont un médicament dérivé du sang qui est utilisé en cas de forme sévère pour l’induction de la rémission.
Les traitements immunomodulateurs de fond (d’épargne cortisonique et d’épargne en immunoglobulines intraveineuses) sont systématiquement prescrits en même temps que la corticothérapie pour permettre son arrêt le plus tôt possible tout en maintenant la rémission.
Ces médicaments comprennent :
o Des immunomodulateurs synthétiques conventionnels :
o Des immunomodulateurs synthétiques biologiques
o Des immunomodulateurs synthétiques ciblés :
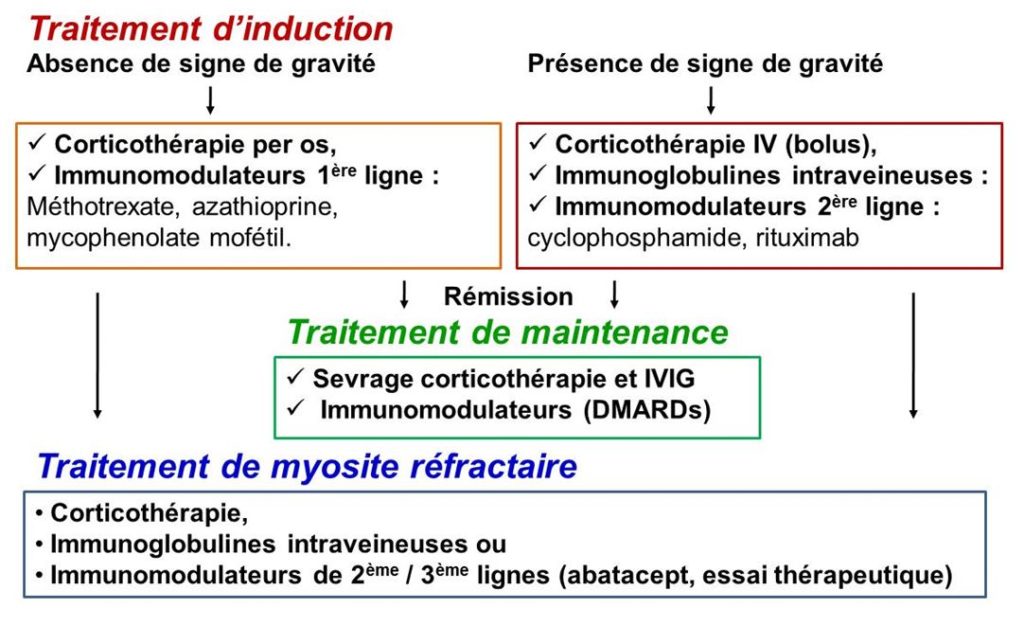
Limiter et reverser les dégâts de la maladie
Les dégâts de la maladie (en opposition a son activité) sont des manifestations résiduelles en lien avec des séquelles et qui ne répondent pas aux immunomodulateurs.
Il s’agit notamment de la sarcopénie, la dépigmentation cutanée, la calcinose.
L’instauration précoce du traitement et son adaptation limite le risque de dégâts.
Les programmes de réentrainement physique sont efficaces sur les dégâts musculaires et doivent être systématiques. La chirurgie permet de traiter la calcinose. La myotomie ou l’injection de toxine botulique dans le muscle crico- pharyngé est utile pour prendre en charge la dysphagie au cours des myosites à inclusions.
Prévenir les co-morbidités
Les risque cardiovasculaire, thromboembolique, infectieux et fracturaire sont augmentés au cours des myosites. Ils doivent être évalués et limités selon les recommandations en vigueur.
Les statines ne sont pas contre indiquées et doivent être prescrites en suivant les recommandations spécifique à la prévention du risque cardiovasculaire.
Autres mesures.
o Traitements spécifiques (RGO, syndrome de Raynaud, HTAP),
o Contraception,
o Photoprotection en cas de dermatomyosite,
o Education thérapeutique,
o Association de patients,
o Ergothérapie,
o Veiller à l’insertion sociale et professionnelle,
o Plusieurs essais thérapeutiques sont en cours au CNR
LES MYOPATHIES INFLAMMATOIRES (MYOSITES)
LES CAUSES DES MYOSITES
o Immunogénétique : les myosites ne sont pas des myopathies génétiques. Cependant, des variants (polymorphismes) génétiques dans des gènes codant des protéines de l’immunité ont été reliés à chaque sous-groupe
o Cancer et myosites : le lien entre cancer et myosite décrit plus haut indique un rôle causal du cancer dans certaines formes de myosite.
o Radiations ultraviolettes et les dermatomyosites : l’incidence de certaines dermatomyosites a été reliée au rayonnement ultraviolet.
o Immunité antivirale et dermatomyosites : le rôle de l’environnement viral au cours des dermatomyosites est suggéré par le fait que la notion d’une infection virale.
o Médicaments : certains médicaments ont été associés à la survenue de myosites. Les mieux caractérisés sont les statines. En France, environ la moitié des patients atteints de myosites nécrosante avec anticorps anti-HMGCR ont pris des statines.
o Tabac et syndrome des antisynthétases : le tabagisme est plus fréquent chez les patients porteurs d’anticorps anti-Jo1
Plusieurs projets de recherches sont en cours au centre de référence de Strasbourg pour mieux comprendre l’origine des myosites.