
Focus sur une pathologie
CONNECTIVITE MIXTE (syndrome de sharp)
La connectivite mixte (parfois appelé syndrome de Sharp) est une forme de maladie auto-immune systémique à la frontière de plusieurs autres pathologies telles que le lupus érythémateux systémique, la sclérodermie systémique, les myosites inflammatoires ou bien encore la polyarthrite rhumatoïde auxquelles elle empreinte une ou plusieurs caractéristiques sans complètement en remplir tous les critères. La maladie est caractérisée par la présence d’un ou plusieurs auto-anticorps, en particulier les anti-RNP (ribonucléoprotéine). Parfois définie comme une forme « d’attente » avant l’évolution vers une autre maladie systémique comme le lupus érythémateux systémique, une proportion importante des patients atteints de cette maladie n’évoluera cependant pas vers une autre maladie auto-immune systémique et certaines caractéristiques cliniques telles que l’atteinte pulmonaire lui sont plus spécifique.
CONNECTIVITE MIXTE
Epidemiologie
La connectivite mixte est une pathologie rare. Sa prévalence est estimée entre 0.21 et 10 pour 100 000 habitants selon les régions du globe. Cette prévalence reste incertaine en France et dans le monde du fait des divergences liées aux critères de classification de la maladie et la confusion fréquente entre connectivite mixte, syndrome de chevauchement et connectivite indifférenciée. Cette confusion tient à la traduction imparfaite du terme MCTD « Mixed Connective Tissue Disease » utilisé dans la littérature. Il existe une prédominance féminine. La pathologie semble débuter plus fréquemment autour de l’âge de 30 ans mais peut se manifester à tout âge.
CONNECTIVITE MIXTE
MANIFESTATIONS CLINIQUES
Quels sont les symptomes de la connectivite mixte?
On estime que 25 à 50% des patients qui se présentent dans un service de référence des maladies auto-immunes pour des symptômes de maladie systémique ne remplit pas les critères d’une seule maladie auto-immune. Parmi ces patients, certains vont développer dans les semaines ou les mois qui suivent une maladie auto-immune caractérisée (Lupus, Sclérodermie, Myosite inflammatoire, PR…), d’autres vont conserver des symptômes partagés avec plusieurs maladies auto-immunes (connectivite indifférenciée s’ils ne remplissent les critères d’aucune maladie de système ou overlap syndrome s’ils remplissent les critères de plusieurs maladies auto-immunes) ou évoluer vers un tableau de connectivite mixte avec la présence d’anticorps anti-RNP (véritable « signature » de la maladie).
Le tableau clinique de connectivite mixte est souvent incomplet au diagnostic et s’étoffe au cours de l’évolution de la pathologie. Initialement, les premiers symptômes peuvent être peu spécifiques et associent (le plus fréquemment) des myalgies, des arthralgies, un syndrome de Raynaud et des œdèmes des mains. La pathologie va associer des traits cliniques de plusieurs maladies auto-immunes, à savoir le lupus érythémateux systémique, la sclérodermie systémique, la polymyosite et la polyarthrite rhumatoïde. Parfois, la maladie peut évoluer vers l’une de ces pathologies.
Manifestations articulaires :
L’atteinte articulaire est fréquente dans la connectivite mixte (60 à 100 % des patients). Il peut s’agir aussi bien d’arthralgies que d’arthrites des petites et grosses articulations, pouvant évoluer vers des lésions érosives et destructrices. Le plus fréquemment, l’atteinte articulaire sera proche de celle observée dans la polyarthrite rhumatoïde avec une atteinte symétrique, polyarticulaires, des petites articulations des mains et des pieds. L’évolution de l’atteinte articulaire dans la connectivite mixte semble moins sévère que dans la polyarthrite rhumatoïde érosive, mais plus sévère que dans le lupus ou la sclérodermie systémique. L’atteinte articulaire de la connectivite mixte peut aussi prendre l’aspect d’une main de Jaccoud ou de main boudinée (« puffy-hand »).
Manifestations musculaires :
Les manifestations musculaires sont fréquentes mais leur prévalence variable selon les études. Il existe le plus fréquemment des myalgies, plus ou moins associées à une faiblesse musculaire, prédominant au niveau des muscles proximaux.
Manifestations vasculaires :
La majorité des patients présente un phénomène de Raynaud, qui peut précéder les autres manifestations cliniques de plusieurs mois voire années. Les anomalies capillaroscopiques sont proches de celles observées dans la sclérodermie systémique avec la présence de mégacapillaires, de micro-hémorragies, d’une raréfaction capillaire… Le syndrome de Raynaud peut se compliquer d’ulcérations digitales.
L’une des complications les plus sévères rencontrée dans la connectivite mixte est l’hypertension artérielle pulmonaire (HTAP). Elle peut être dépistée par l’existence de signes cliniques (fatigue, malaise, dyspnée, souffle d’insuffisance tricuspidienne se majorant à l’inspiration profonde (signe de Carvallo), éclat du B2 au foyer pulmonaire) ; dépistée par échocardiographie (ETT) ; mais elle est affirmée par le cathéterisme cardiaque droit. L’HTAP se caractérise par une pression artérielle pulmonaire moyenne supérieure à 25 mmHg au repos avec une pression capillaire inférieure à 15 mmHg. Elle se développe le plus souvent durant l’évolution de la maladie et est retrouvée chez 20 à 30 % des patients. C’est la principale cause de décès dans cette pathologie. Elle nécessite un dépistage régulier par échographie trans-thoracique, épreuves fonctionnelles respiratoires et dosage du BNP ou NT-pro-BNP.
Manifestations pulmonaires :
Les atteintes pulmonaires à type de pneumopathie interstitielle (NSIP ou UIP) sont fréquentes dans les connectivites mixtes (60-70%) et font l’une des spécificités de la pathologie. Elles peuvent s’associer à une baisse de la DLCO aux épreuves fonctionnelles respiratoires et peuvent évoluer vers la fibrose qui font toute la gravité de cette atteinte. La majorité des patients est cependant peu symptomatique.
Une atteinte pleurale peut aussi se rencontrer avec des épanchements pleuraux bilatéraux, souvent peu symptomatiques.
Manifestations cutanées
Elles se caractérisent par des manifestations cutanées typiques de la sclérodermie systémique ou du lupus érythémateux systémiques. L’œdème des mains et des doigts (« puffy hands ») est une manifestation fréquente et souvent présente au diagnostic. La sclérodactylie, la calcinose cutanée, les télangiectasies peuvent également se rencontrer. Les manifestations lupus-like incluent le rash malaire, la photosensibilité et le lupus discoïde. Enfin, des traits cliniques du syndrome de Goujerot-Sjögren peuvent être observés avec notamment une sècheresse ophtalmologique et buccale.
Manifestations gastro-intestinales
La principale atteinte digestive est le trouble de la motilité œsophagienne, se traduisant cliniquement par un reflux gastro-œsophagien.
Manifestations cardiaques
L’atteinte péricardique (péricardite) est la plus fréquemment rencontrée. Un risque athérogène et cardiovasculaire augmenté est également rapporté.
Manifestations hématologiques
Des cytopénies sont fréquemment retrouvées (anémie, thrombopénie, lymphopénie). Le test de Coombs est rarement positif.
Il est a noter que les atteintes rénales et du système nerveux central sévères sont extrêmement rare dans la connectivite mixte. Leur présence doit faire réévaluer le diagnostic ou suspecter une évolution vers une autre maladie (Lupus érythémateux systémique ou Sclérodermie systémique).
CONNECTIVITE MIXTE
DIAGNOSTIC
Tous les patients atteints de connectivite mixte ont des anticorps anti-nucléaires positifs (fluorescence mouchetée à gros grains irréguliers) avec une spécificité anti U1-snRNP. La méthode de détection et le titre d’anticorps nécessaire à leur positivité n’est cependant pas défini de manière consensuelle. Le titre de ces auto-anticorps pourrait être corrélé à l’activité de la maladie.
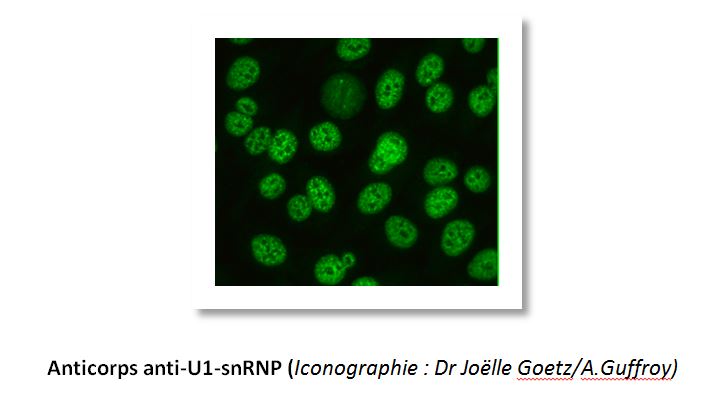
Un grand nombre de patients auront également un facteur rhumatoïde positif ou des anticorps anti-phospholipides. L’hypergammaglobulinémie est également fréquente. Une cryoglobuline et une hypocomplémentémie peuvent également se rencontrer.
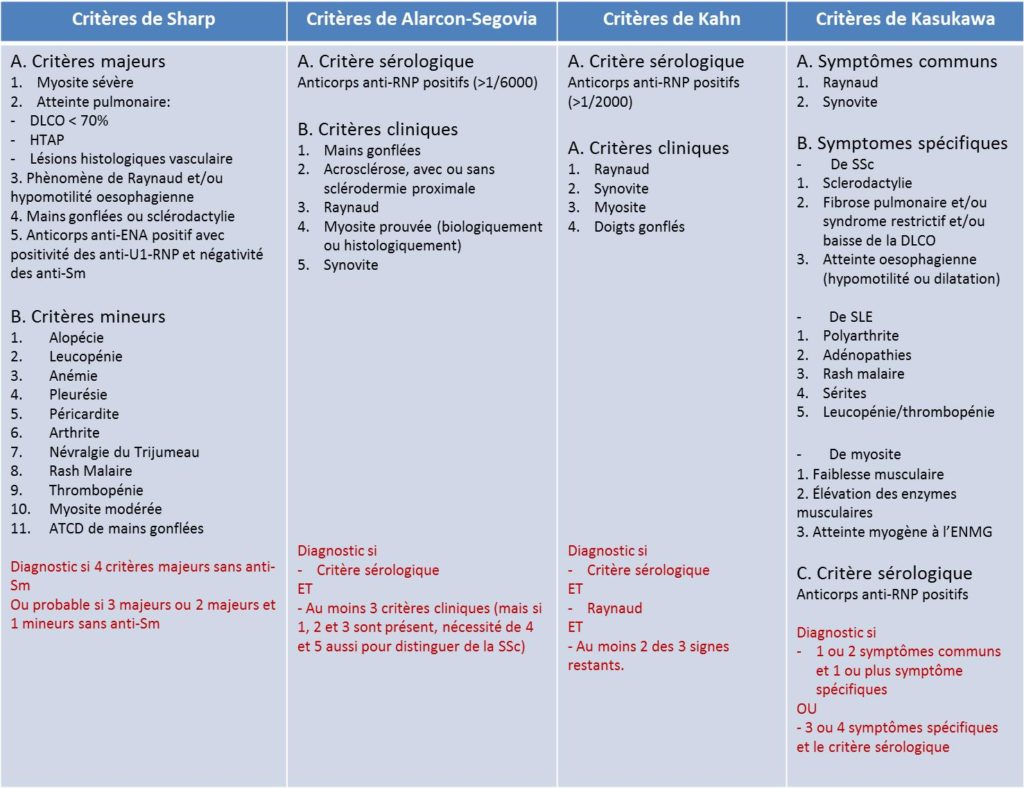
Quatre classifications diagnostiques différentes, à savoir les classifications de Sharp, Alarcon-Segovia, Kahn et Kasukawa, ont été proposées sans qu’aucune ne soit clairement supérieure en terme de sensibilité et spécificité du diagnostic. Ces classifications incluent les symptômes cliniques les plus fréquents et la positivité des anticorps anti-RNP.
CONNECTIVITE MIXTE
PATHOGENIE
La physiopathologie de la maladie est partiellement connue à l’heure actuelle. Sur un terrain génétique prédisposé, l’émergence d’auto-anticorps anti-ribonucléoprotéine (comme l’anti-U1-RNP) vont former des complexes immuns qui vont s’accumuler et favoriser l’activation du système immunitaire inné via les récepteurs de danger (Toll-Like Receptor, TLRs). Il s’en suit une boucle d’activation du système immunitaire adaptatif (lymphocytes T et lymphocytes B) avec la production de cytokines pro-inflammatoires (comme l’interféron de type I) qui vont être à l’origine des lésions tissulaires constatées. Ce schéma physiopathologique est proche de celui du lupus érythémateux systémique.
CONNECTIVITE MIXTE
TRAITEMENTS
La prise en charge thérapeutique de cette maladie rare et polymorphe nécessite une approche pluridisciplinaire et coordonnée entre la ville et l’hôpital, le médecin référent et le médecin traitant et différents professionnels de santé (kinésithérapeute, infirmières d’éducation thérapeutiques, diététiciennes, assistante sociale) en fonction du profil évolutif de chaque patient.
L’évaluation du risque cardiovasculaire à long terme et l’individualisation vers une autre maladie systémique (LES, Sclérodermie) seront systématiquement recherchés pour éviter les complications propres à ces pathologies.
Les atteintes articulaires peuvent être traitées symptomatiquement par AINS, s’il s’agit d’arthralgies ou d’arthrites non érosives. En cas d’inefficacité, de contre-indications aux AINS ou de présence d’érosions, un traitement par Methotrexate en première intention semble le meilleur choix plus ou moins associé à une corticothérapie. Si ce traitement s’avère insuffisant, une biothérapie par anti-TNF alpha peut être envisagée.
La présence d’une myosite inflammatoire justifie l’utilisation de corticoïdes plus ou moins associé à de l’Azathioprine, du Methotrexate et/ou des immunoglobulines intraveineuses en fonction du degré d’atteinte.
Le traitement du syndrome de Raynaud est dans un premier temps symptomatique avec l’éviction du froid, le port de gant, l’arrêt du tabagisme, la contre-indication aux traitements vasoconstricteurs périphériques tels que les béta bloquants. Les inhibiteurs calciques sont la première ligne du traitement médical. Des perfusions d’Ilomédine peuvent être envisagées en cas d’ulcérations sévères ou de gangrène. Le Bosentan peut être utilisé pour réduire le nombre d’ulcères digitaux chez les patients présentant des ulcérations à répétition.
Les traitements spécifiques de l’HTAP incluent les antagonistes du récepteur à l’endothéline 1 tel que le Bosentan, les inhibiteurs de la phosphodiesterase tels que le Sildenafil et les prostaglandines tels que l’Epoprostenol ou la Prostacycline. Les inhibiteurs calciques de longue durée d’action peuvent également être indiqués. Certains patients pourraient répondre à un traitement immunosuppresseur par Corticoïdes et Cyclophosphamide.
Les atteintes pulmonaires peuvent être traitées par corticothérapie et peuvent requérir en cas d’atteinte sévère et/ou évolutive un traitement par Cyclophosphamide.
Les patients présentant des manifestations cutanées lupus-like doivent éviter l’exposition solaire et utiliser les crèmes solaires. Une corticothérapie et un traitement par Plaquenil peuvent être nécessaires si la photoprotection est inefficace.
Dans les formes sévères ou réfractaires, un traitement par intensification thérapeutique/greffe de cellules souches hématopoïétiques (thérapie cellulaire) peut être envisagé et discuté en Réunion de Concernation Pluridisciplinaire (RCP MATHEC) nationale bien que l’on dispose de peu de données objectives sur son efficacité dans cette indication du fait du peu de malades traités.